Finally, we can use the new FDA CCP to eliminate FedEx shipments, and 100% of your submissions will be electronic through the portal.
If you need help preparing your FDA eCopy submission, contact Medical Device Academy.
July 2022 Update for the FDA eCopy process
The FDA created a Customer Collaboration Portal (CCP) for medical device manufacturers. Originally, the portal’s purpose was to provide a place where submitters can track the status of their submissions and verify the deadlines for each stage of the submission review process. Last week, on July 19, the FDA emailed all active FDA CCP account holders that they can upload both FDA eCopy and FDA eSTAR files to the portal 100% electronically. Since our consulting team sends out submissions daily, everyone on the team was able to test the new process. If you have a CCP account, you no longer need to ship submissions via FedEx to the Document Control Center (DCC).
FDA CCP step-by-step uploading process
When you are uploading an FDA eCopy for medical device submission to the Document Control Center (DCC), using the new FDA CCP, the following steps are involved:
- Confirm your eCopy complies with FDA’s eCopy guidance.
- Compress your eCopy into a “.zip” file.
- Sign in to the portal on the login page
- Click on the “+” symbol on the left panel of the webpage (if you hover over the “+” symbol, you will see “Send a submission”)
- Select your desired upload format (pre-submissions, meeting minutes, breakthrough device designations, and withdrawal letters must be submitted as an eCopy)
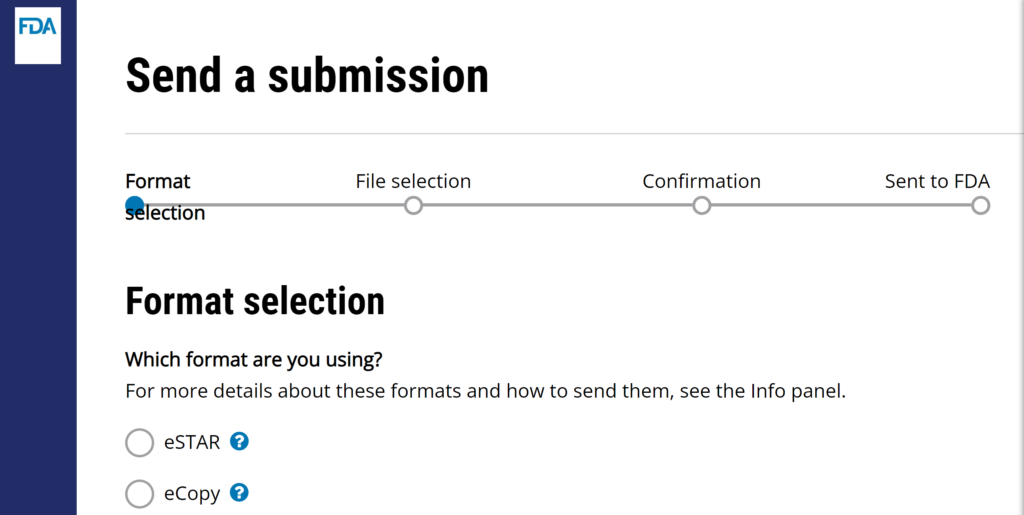
- Click on the “Next” button that appears below the selection formats once a format is selected
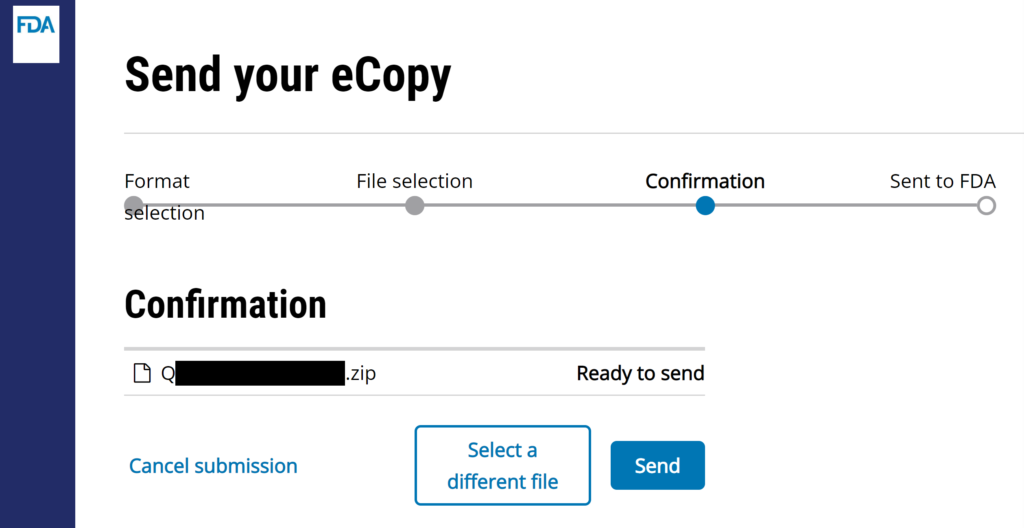
- Drag & drop your single “.zip” file here, or browse for it.
- Click on “Send” button to complete the uploading process.
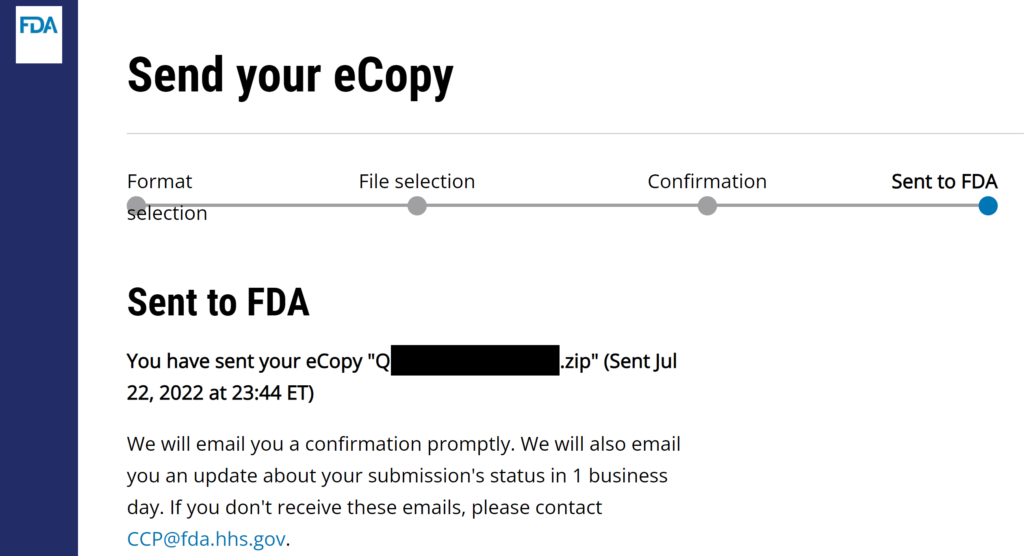
- Verify that the FDA CCP site gives you a confirmation for the successful uploading of your submission.
FDA Q&A about the new FDA CCP Submission Uploading Process
- Medical Device Academy Question: Who will be permitted to use the FDA CCP to upload submissions for the DCC? FDA Response: We will first offer this feature in batches to people like you who already use CCP so we can study its performance. We will then refine it and make it available to all premarket submitters.
- Medical Device Academy Question: What do you need to use the FDA CCP? FDA Response: You don’t need to do anything to participate since you already use CCP. We will email you again when you can start sending your next submissions online.
- Medical Device Academy Question: Suppose another consultant asks me to submit an eSTAR or eCopy for them, or I do this for a member of my consulting team. Is there any reason I cannot upload the submission using my account even though the other person is the official submission correspondent and their name is listed on the cover letter? FDA Response: The applicant and correspondent information of the submission is still used when logging the submission in. The submitter (i.e., the person uploading the submission) is not used in any part of the log-in process. The submission portal is essentially replacing snail mail only; once the DCC loads the submission, whether it be from a CD or an online source, the subsequent process is identical to what it used to be, for now.
- Medical Device Academy Question: Is there any type of eCopy that would not be appropriate for this electronic submission process (e.g., withdrawal letters, MAF, or breakthrough device designations)? FDA Response: You can use the eCopy option to submit anything that goes to the DCC, so all your examples are fair game, though interactive review responses would still be emailed to the reviewer.
- Medical Device Academy Question: How can I get help from the FDA? FDA Response: If you have questions, contact us at CCP@fda.hhs.gov.